Beneath your skin, a cell is rusting. Brown-yellow stains creep across its membrane. Pop—the whole sheet tears open, splashing pro-inflammatory debris onto its neighbors. That is ferroptosis. The cell next door dies differently: a protein punches holes through the membrane, the cell swells and bursts—like a micro-grenade. That is pyroptosis. Four studies in 2026 converge on a single finding: these two "explosive" deaths can lock aging and inflammation into one loop—and that loop has brakes.
Why the way a cell dies shapes how fast you age
Billions of your cells die every day. Most go quietly, like autumn leaves drifting down, swept up by patrolling immune cells—a process called apoptosis. Ferroptosis and pyroptosis are different: they die like water balloons, membranes rupturing, contents splattering, dumping pro-inflammatory cargo into the surrounding tissue.
Neighboring cells, drenched in this "chemical splash," are pushed toward premature senescence. Senescent cells neither die nor leave; they just keep secreting inflammatory signals (scientists call this SASP). Those signals, in turn, push yet more cells toward rupturing death. The loop starts turning: death → inflammation → senescence → more death.
More than 1.4 billion people worldwide are now over 60. Chronic inflammation is the common backdrop of nearly every disease of aging. The question gets concrete: can we intercept the loop at the point of "how the cell dies"? Four studies in 2026—spanning skin, brain, and kidney—offer different facets of the same answer.
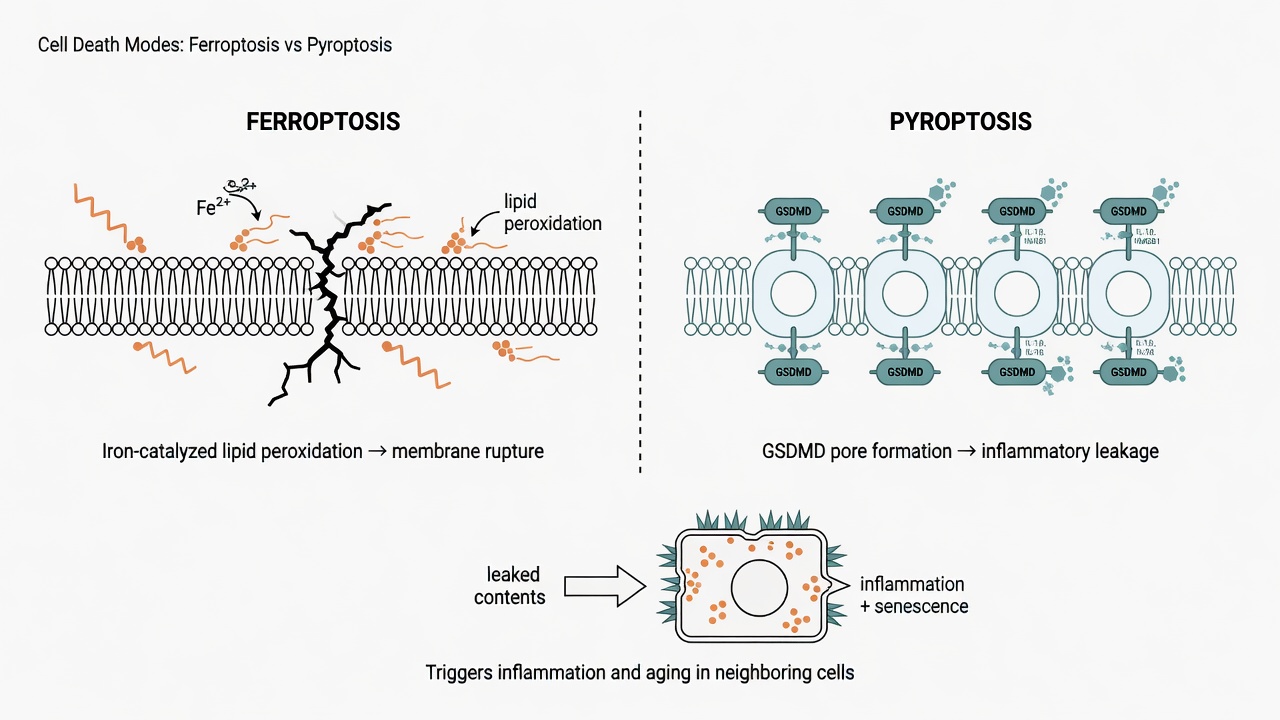 Figure 1: Two "rupturing" deaths—lipid peroxidation rusts the membrane apart (ferroptosis), or gasdermin punches holes through it (pyroptosis). Both splash pro-inflammatory cargo onto neighbors.
Figure 1: Two "rupturing" deaths—lipid peroxidation rusts the membrane apart (ferroptosis), or gasdermin punches holes through it (pyroptosis). Both splash pro-inflammatory cargo onto neighbors.
A rusting suicide in the skin: how ferroptosis breeds senescent cells
Your dermis is home to legions of fibroblasts—the cells that build collagen and hold your skin together. Under ferroptotic stress, they stumble onto a fork: they head toward death and toward senescence at the same time. The two bad outcomes are not coincidental; they are threaded onto the same signaling axis.
In 2026, Ng and colleagues found the valve on that axis—hypoxia-inducible factor HIF-1α. It damps the NF-κB–DPP4 pathway, letting fibroblasts dodge both ferroptosis and senescence features simultaneously. The core of ferroptosis is lipid peroxidation: iron-catalyzed oxidation rusts the membrane's fatty acids, like a seaside iron gate corroding in the salt wind, until the whole panel collapses.
When NF-κB ramps DPP4 up, fibroblasts become more prone to ferroptosis and more likely to enter senescence. Put differently, the senescent cells that linger in your aging skin—smoldering with inflammation—may have been pushed there by ferroptotic stress all along. Loosen the HIF-1α valve and both bad outcomes recede.
A self-spreading alarm in the brain: the two faces of pyroptosis
The most dangerous thing about pyroptosis is not that it kills a cell—it is that it relays itself.
In a 2026 brain-injury model, Li and colleagues traced a self-perpetuating circuit. When neurons undergo pyroptosis, the pore-forming protein GSDMD releases mitochondrial DNA (mtDNA). That stray mtDNA drifts like a lit match into the AIM2 inflammasome, igniting more neuronal pyroptosis and dragging cognition down with it. The product of death becomes the fuse for the next round.
A brake was found in the brain too. Xu and colleagues showed that TSG-6, secreted by bone-marrow mesenchymal stem cells, can modulate NLRP3 inflammasome activation in microglia—the brain's immune patrol officers, responsible for engulfing debris and monitoring threats. If the patrol officers themselves catch fire from pyroptosis, the blaze only spreads. TSG-6 acts like fireproof gear for those officers.
Read the neurons' relay fire alongside the microglia's role as fire-source manager, and two actionable intervention points emerge: block the mtDNA–AIM2 baton pass, or calm overactivated patrol officers.
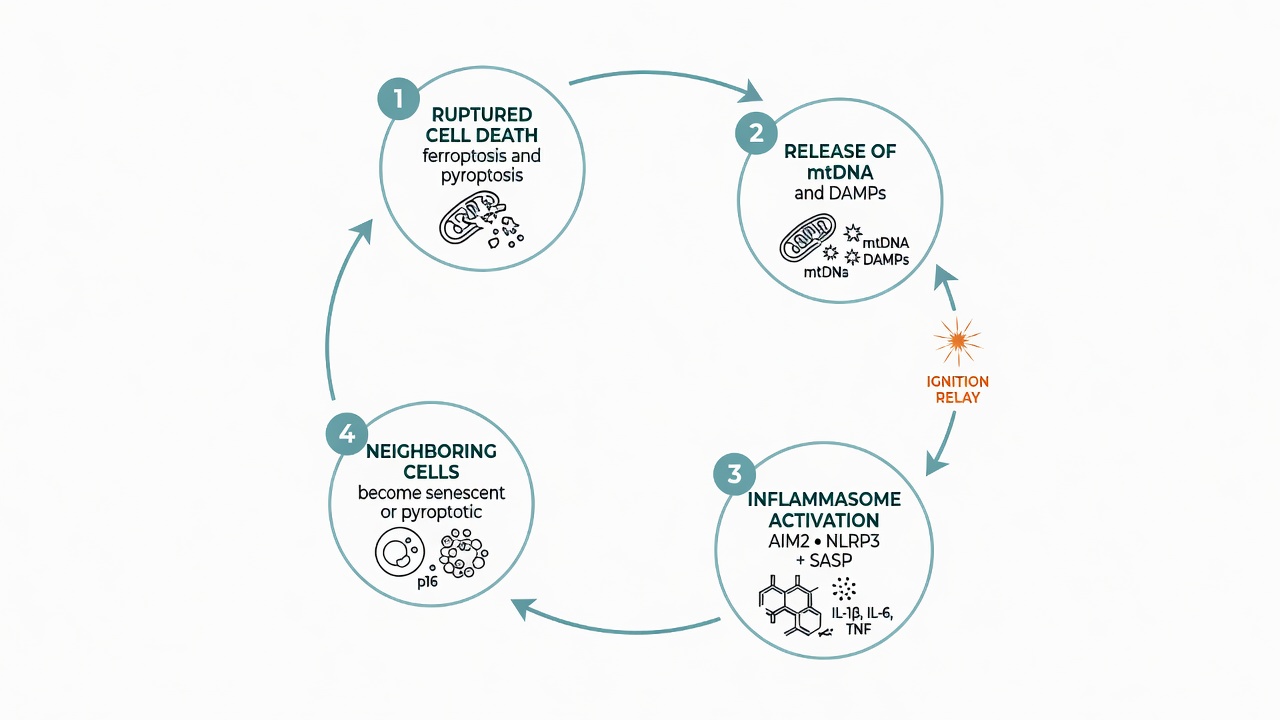 Figure 2: The self-spreading loop—rupturing death releases mtDNA and pro-inflammatory cargo, activating inflammasomes and SASP, pushing neighboring cells into senescence or pyroptosis, and reigniting more inflammation.
Figure 2: The self-spreading loop—rupturing death releases mtDNA and pro-inflammatory cargo, activating inflammasomes and SASP, pushing neighboring cells into senescence or pyroptosis, and reigniting more inflammation.
How to tap the brakes—and why you cannot slam them
The goal is not to abolish cell death but to turn down the volume on the rupturing kind. In a diabetic-nephropathy model, Sun and colleagues found that Pueraria lobata extract suppresses the NLRP3/Caspase-1 pathway, easing pyroptosis and inflammation in the kidney. Caspase-1 is one of pyroptosis's executioners; the extract effectively targets the executioner's wrist, handing natural-product intervention a precise molecular target.
But you may have already spotted a flip side: pyroptosis and ferroptosis are normal weapons of the immune system. Pyroptosis clears infected cells; a measured amount of cell death is part of anticancer defense. Shut these pathways down entirely and you disarm the immune system. As inflammasome inhibitors like MCC950 move toward clinical trials, dose windows and organ selectivity are what researchers worry about most. The deeper complication: the same pathway can play opposite roles in different organs—braking pyroptosis in the kidney may help, but in a tumor microenvironment it could let cancer cells slip past immune surveillance. A one-drug-fits-all approach is nearly impossible. Future strategies will likely target specific tissues and time windows.
As of 2026, at least four molecular pathways have been shown to regulate the "rupturing death → senescence" loop: HIF-1α–NF-κB–DPP4 (skin ferroptosis), GSDMD–mtDNA–AIM2 (neuronal pyroptosis relay), TSG-6–NLRP3 (microglial pyroptosis), and puerarin–Caspase-1 (renal pyroptosis). According to WHO, the global population over 60 has reached 1.4 billion and is projected to double to 2.1 billion by 2050. Chronic low-grade inflammation (inflammaging) is the shared substrate of cardiovascular disease, neurodegeneration, and type 2 diabetes. Ferroptosis and pyroptosis, as upstream drivers of inflammaging, are emerging as the second major anti-aging intervention track after senolytics.
These four studies remain largely in animal and cell models. Whether the benefits translate safely to humans, and at what dose windows, awaits clinical trials. The direction is clear: rather than waiting until senescent cells pile up, we can turn the inflammatory loop down at the very moment a cell decides how to die—like adjusting a thermostat, not ripping out the furnace.
You might also wonder
Q: How are ferroptosis and pyroptosis different from ordinary apoptosis?
Apoptosis is a quiet demolition job: the cell cuts itself into tidy parcels, which immune cells sweep away without triggering inflammation. Ferroptosis and pyroptosis are "explosive" deaths—the membrane ruptures, contents leak out, and the surrounding tissue is drenched in pro-inflammatory material. It is precisely this "dying loudly" quality that makes them a bridge between aging and chronic inflammation. All three are forms of regulated cell death; the difference lies in how the cell dies and what happens to its neighbors.
Q: Since ferroptosis is linked to aging, can I fight aging by cutting iron intake or loading up on antioxidants?
That leap is not supported. The key to ferroptosis is not "too much iron in the body" but a breakdown of lipid-peroxidation defenses inside specific cells. Indiscriminate iron restriction can cause anemia, and high-dose antioxidant supplements have not been shown in clinical trials to slow aging. Current research targets intracellular signaling axes (e.g., HIF-1α–DPP4), not systemic nutrient adjustment. Until molecular mechanisms translate into clinical recommendations, balanced nutrition and regular check-ups remain the practical advice.
References
- Ng et al. (2026). HIF-1α attenuates ferroptosis-associated dermal fibroblast senescence via modulation of NF-κB–DPP4 signaling. Journal of Translational Medicine. doi: 10.1186/s12967-026-08445-y
- Li et al. (2026). A self-perpetuating neuron-intrinsic GSDMD–mtDNA–AIM2 inflammasome axis drives neuronal pyroptosis and cognitive impairment after traumatic brain injury. Frontiers in Immunology. doi: 10.3389/fimmu.2026.1867920
- Xu et al. (2026). Bone marrow mesenchymal stem cell-derived TSG-6 regulates microglial pyroptosis via NLRP3 inflammasome. Stem Cell Research & Therapy. doi: 10.1186/s13287-026-05121-2
- Sun et al. (2026). Pueraria lobata extract mitigates diabetic nephropathy via NLRP3 inflammasome and Caspase-1 regulation. Scientific Reports. doi: 10.1038/s41598-026-57815-5
Frequently Asked Questions
鐵死亡和焦亡,跟一般的「細胞凋亡」有什麼不同?
凋亡是「乾淨退場」——細胞縮小、被打包回收,幾乎不漏內容物。鐵死亡(膜被氧化崩解)與焦亡(gasdermin 在膜上打洞)則會破裂、把促發炎物質潑向鄰居,因此更容易點燃發炎與衰老。
所以把鐵死亡和焦亡完全抑制掉,就能抗老?
不是。這是最常見的誤解。焦亡是免疫系統清除受感染細胞的正常武器,適度的細胞死亡也是組織更新與抗癌防線。長期、全面關閉這些路徑,可能削弱對感染與癌變的防禦。研究的目標是把過度活化的迴圈「調回平衡」,不是按下總開關。
這些研究能直接套用在人身上嗎?
目前還不行。本文四項研究多在動物與細胞模型完成,療效與安全劑量範圍仍待人體臨床試驗確認。文中的分子靶點(HIF-1α、TSG-6、Caspase-1)是「可介入的方向」,而非已驗證的療法。
葛根萃取物可以拿來保養腎臟嗎?
這項結果來自糖尿病腎病的動物模型,顯示葛根能調控 NLRP3/Caspase-1 路徑,但劑量、純度與人體效果都未經臨床確認。請勿自行把實驗結果當成保健建議,用藥或保健品請先諮詢專業人員。
既然鐵死亡跟衰老有關,減少鐵攝取或大量補充抗氧化劑就能抗老嗎?
不能這樣簡單推論。鐵死亡的關鍵不是「體內鐵太多」,而是特定細胞內脂質過氧化防禦失衡。盲目減鐵可能造成貧血,大量補充抗氧化劑也未在臨床試驗中證實能延緩衰老。研究鎖定的是細胞內特定訊號軸(如 HIF-1α–DPP4),不是全身性營養素調整。
Found this useful?
Follow for new AI × biomedical research notes:
Or buy me a coffee to keep new content coming.
☕ Buy Me a Coffee