Older bodies often show elevated inflammatory markers in blood without any infection or injury. This invisible, low-grade, chronic burn is called inflammaging. It is not a passive side effect of getting weaker — it has a concrete ignition switch.
Why does aging come bundled with inflammation that never quite shuts off? Three 2026 studies point to the same culprit: the innate immune system's alarm sensors. Aging cells leak nucleic acids that should stay locked in the nucleus, and those sensors mistake them for a virus. Aging also jams immune cells into a trigger-happy state. The encouraging part: the same sensors that fire the switch are also where a brake can be applied.
Why does an aging body inflame without an enemy?
At its core, inflammaging is a false alarm. Senescent cells emit signals that look like infection, so the immune system keeps responding to an enemy that never arrives. That idling inflammation is the shared soil beneath diabetes, cardiovascular disease, dementia, and frailty.
Inflammation is meant to work like an emergency room: detect an intruder or injury, put out the fire, repair, then power down. Aging breaks the off-switch. To see where it jams, start with the first spark.
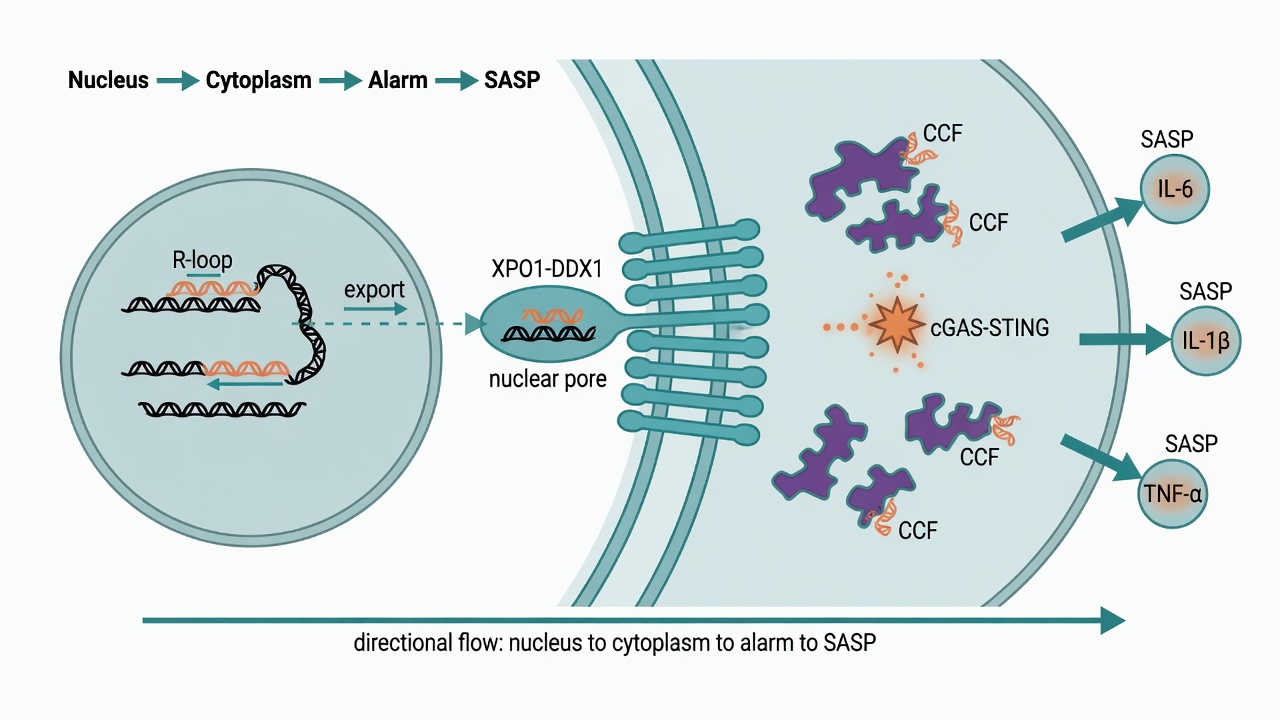
The first spark: nucleic acids leak out of the nucleus
One source of the inflammatory signal is nucleic acids in the wrong place. Hao and colleagues found that senescent cells transport R-loops — three-stranded structures of RNA tangled with DNA — from the nucleus into the cytoplasm, where they trip the cGAS-STING pathway built to detect viruses. That forces the cell to pump out inflammatory factors, the senescence-associated secretory phenotype (SASP).
The key is the courier. The study shows that XPO1 and DDX1 form a complex that exports R-loops out of the nucleus and into cytoplasmic chromatin fragments (CCFs), where the alarm goes off. When the team blocked that route with the XPO1 inhibitor KPT-330, the SASP quieted down, age-associated inflammation eased, and the healthspan of mice extended. In other words, the first spark of inflammaging can be pinched off at its source.
The amplifier: aging locks immune cells in the "fire" position
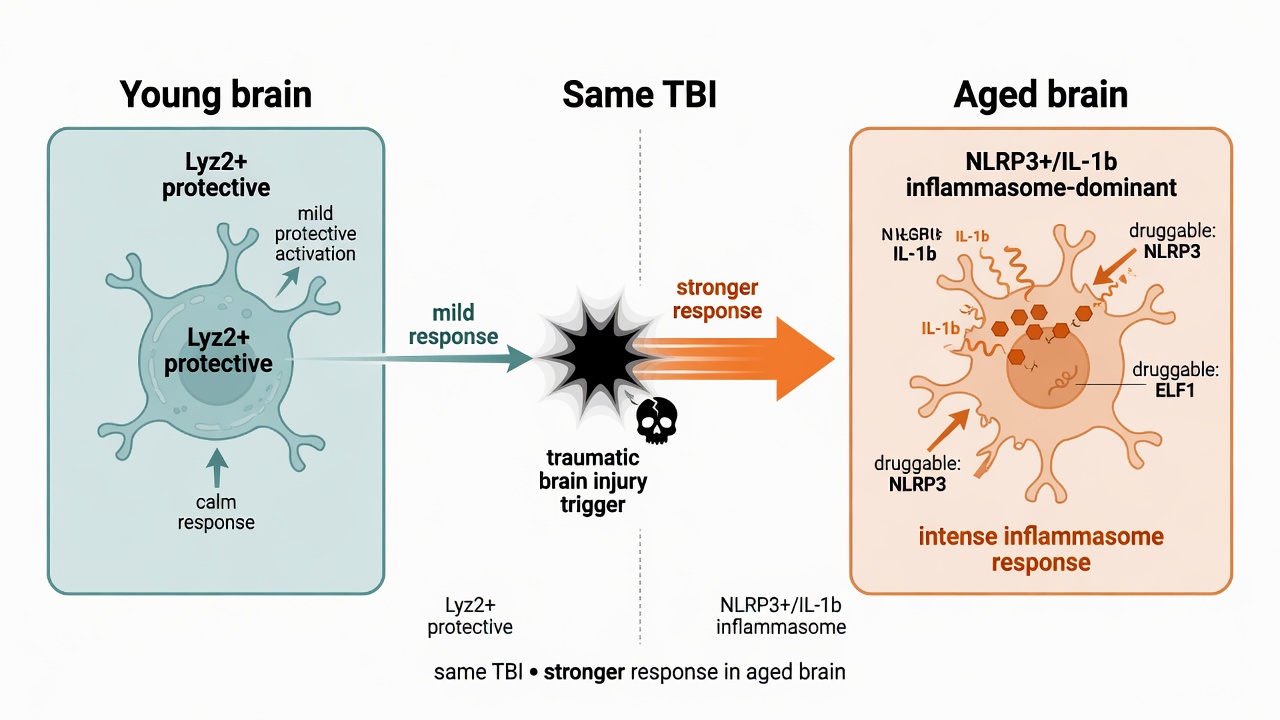
Whether a spark becomes a blaze depends on the state of immune cells — and aging tunes them to the volatile setting. Brain injury is especially deadly for older adults, and a study summarized by Morganti and Bachstetter in the JCI explains the mechanism: aging reprograms the brain's microglia.
In young brains, microglia favor a protective Lyz2+ state; in aged brains, an NLRP3+/IL-1β inflammasome state takes over. The same impact lands harder on aged tissue. By perturbing the NLRP3 and ELF1 switches in mice, researchers shifted the balance back and improved survival; even the repurposed drug Imeglimin helped. That reframes "age" from an unchangeable risk label into a set of druggable cellular targets.
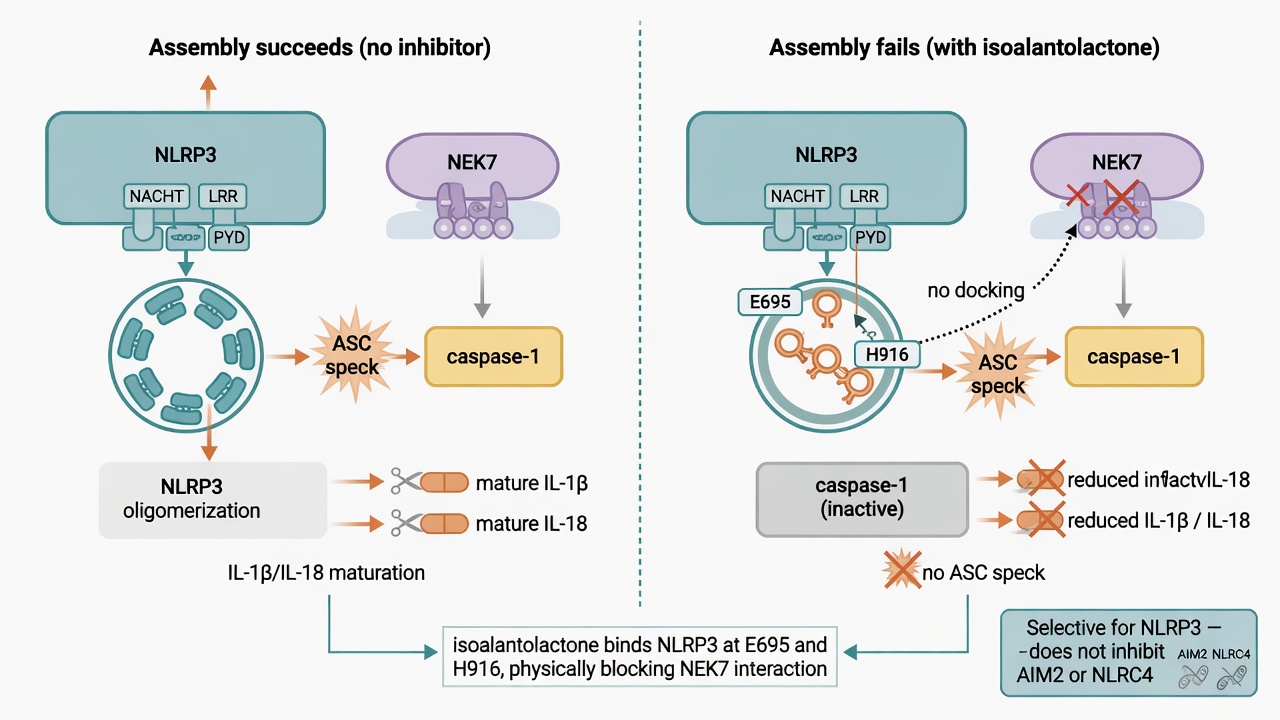
Finding the brake: jamming the inflammasome's assembly
If the NLRP3 inflammasome is the amplifier, can it be jammed directly? Shi and colleagues went after it with a natural product. They found that isoalantolactone, a sesquiterpene lactone, binds NLRP3 directly and disrupts the partnership between NLRP3 and NEK7, so the inflammasome cannot assemble; caspase-1 activation and IL-1β/IL-18 maturation both drop.
Its selectivity is clean: it blocks NLRP3 while barely touching the other two inflammasomes, AIM2 and NLRC4. The team pinned the binding site to two residues on NLRP3, E695 and H916, and saw protection in mouse models of gout and metabolic fatty liver disease — effects that vanished in NLRP3-deficient mice, confirming the drug works through NLRP3. For the old question of whether a natural compound can hit a target precisely, this is a concrete answer.
Don't rush to "switch inflammation off"
It is tempting to stitch the three steps together — pinch the spark, lower the amplifier, hit the brake — and call inflammaging solved. But inflammation is a defense, not pure damage. cGAS-STING and NLRP3 hold day jobs in fighting infection, clearing damaged cells, even tumor surveillance; blanket suppression could open immune gaps.
Three caveats come first. One, the microglia and gout findings are mainly mouse work; whether humans respond the same way awaits clinical data. Two, the XPO1 inhibitor KPT-330 (clinically, selinexor) is a cancer drug with real side effects — using it as an anti-aging agent is a different proposition. Three, senescent cells and inflammasomes are double-edged, with roles in tissue repair and wound healing. "Turn it down" is a more reasonable goal than "shut it off." Seeing inflammaging as a system with switches and brakes — but one you should not rip out entirely — is the real lesson of these three studies.
References
- Hao, X. et al. (2026). Nuclear export of R-loop by the DDX1 and XPO1 complex promotes senescence-associated secretory phenotype and inflammaging. Nature Aging. doi: 10.1038/s43587-026-01147-6
- Morganti, J.M. & Bachstetter, A.D. (2026). Aging reprograms microglia toward an inflammasome-linked response to traumatic brain injury. The Journal of Clinical Investigation. doi: 10.1172/JCI207022
- Shi, Y. et al. (2026). Isoalantolactone targets NLRP3 to disrupt NLRP3-NEK7 interaction and suppress inflammasome activation. Biochemical Pharmacology. doi: 10.1016/j.bcp.2026.118165
Frequently Asked Questions
吃消炎藥能不能對抗發炎老化?
傳統消炎藥壓的是下游症狀,不是上游開關(cGAS-STING、NLRP3 組裝)。目前沒有消炎藥被核准用於抗發炎老化適應症。
吃消炎藥能不能對抗發炎老化?
傳統消炎藥壓的是下游症狀,不是上游開關(cGAS-STING、NLRP3 組裝)。目前沒有消炎藥被核准用於抗發炎老化適應症。
Found this useful?
Follow for new AI × biomedical research notes:
Or buy me a coffee to keep new content coming.
☕ Buy me a coffee