
TL;DR: "Longevity genes" sounds like a lifetime insurance policy. But the latest large-scale genetic analyses are rewriting the script: the same gene that protects you at thirty may turn against you at sixty. Aging isn't a straight downhill slide — it's a tug-of-war played out across time.
Your "Longevity Genes" May Be Betraying You
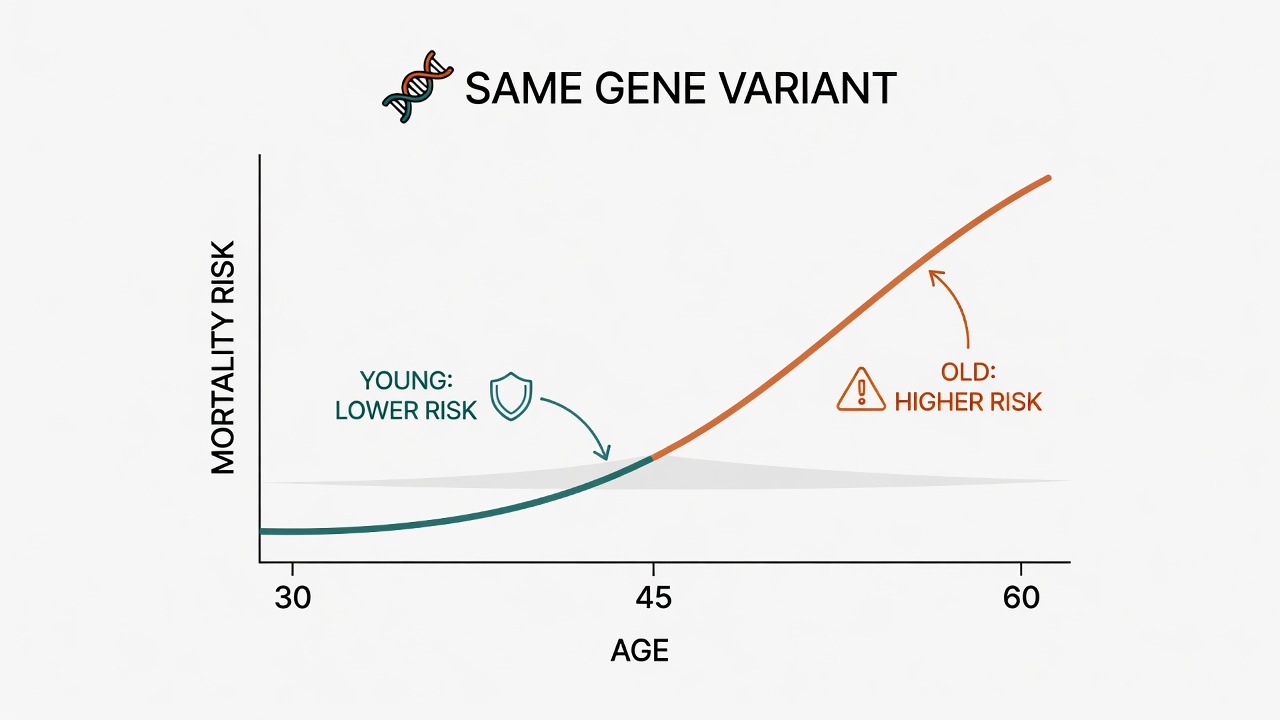
When geneticists stopped looking at "how long you live" as a single endpoint and began dissecting mortality risk by age interval, an unsettling pattern emerged. Many genetic variants don't behave as consistently "good" or "bad." They suppress mortality risk in youth, go quiet in middle age, and then push mortality higher in old age.
This phenomenon has a formal name: antagonistic pleiotropy. Evolutionary biologist George Williams proposed the hypothesis in 1957, but it took large-scale biobanks and sophisticated age-stratified statistical models to produce direct empirical evidence.
Even more counterintuitively, the relationship between BMI and survival isn't fixed either. In younger populations, lower BMI correlates with better survival; with advancing age, the relationship reverses. The same metric points to opposite outcomes at different points in life.
This is not statistical noise. It is a design principle of life itself: natural selection cares about whether you survive to reproductive age and pass on your genes. What happens after seventy is, evolutionarily speaking, not its concern. The genetic armaments that fight for you in your twenties and thirties may become sources of autoimmune-like attack in your sixties and seventies.
Your Body Already Has a Built-In Clock
If gene effects change with time, how does the body "know" what time it is?
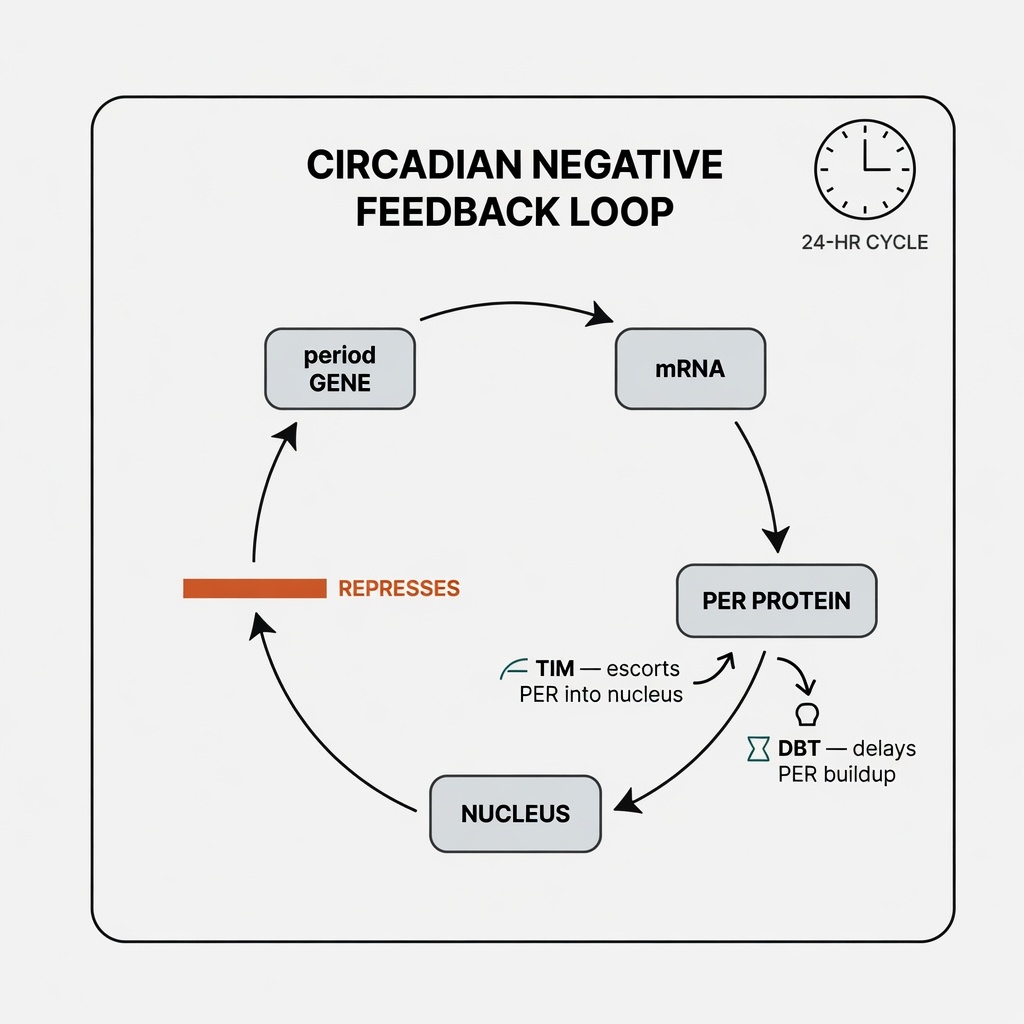
The answer resides in every cell. The 2017 Nobel Prize in Physiology or Medicine was awarded to Jeffrey C. Hall, Michael Rosbash, and Michael W. Young for discovering the molecular mechanisms governing circadian rhythms. Using fruit flies, they isolated the period gene and showed that its product, PER protein, accumulates at night and degrades during the day, forming a precise 24-hour oscillation.
This clock does far more than regulate sleepiness.
PER operates through a negative feedback loop: the period gene is transcribed into mRNA, translated into PER protein, which accumulates until it enters the nucleus and inhibits the gene's own activity. As protein levels fall, the gene reactivates, cycling continuously. Young later identified the timeless gene (encoding TIM protein, which escorts PER into the nucleus) and the doubletime gene (encoding DBT protein, which delays PER accumulation to fine-tune the cycle to approximately 24 hours).
The critical insight: a vast number of genes in your body are regulated by this clock. Hormone secretion, body temperature fluctuations, metabolic rhythms, immune response intensity — all follow circadian patterns. When the clock drifts, the disruption cascades not through a single organ but through the temporal coordination of the entire physiological system.
Aging, in a fundamental sense, is the process of this clock gradually losing its precision.
The First Outpost to Fall: The Neuromuscular Junction
If aging is a tug-of-war across time, who lets go of the rope first?
A systematic review spanning 27 studies points to an answer: the neuromuscular junction (NMJ). The NMJ is the synaptic structure where motor neurons meet skeletal muscle fibers. Every time you raise your hand, walk, or breathe, the signal passes through here.
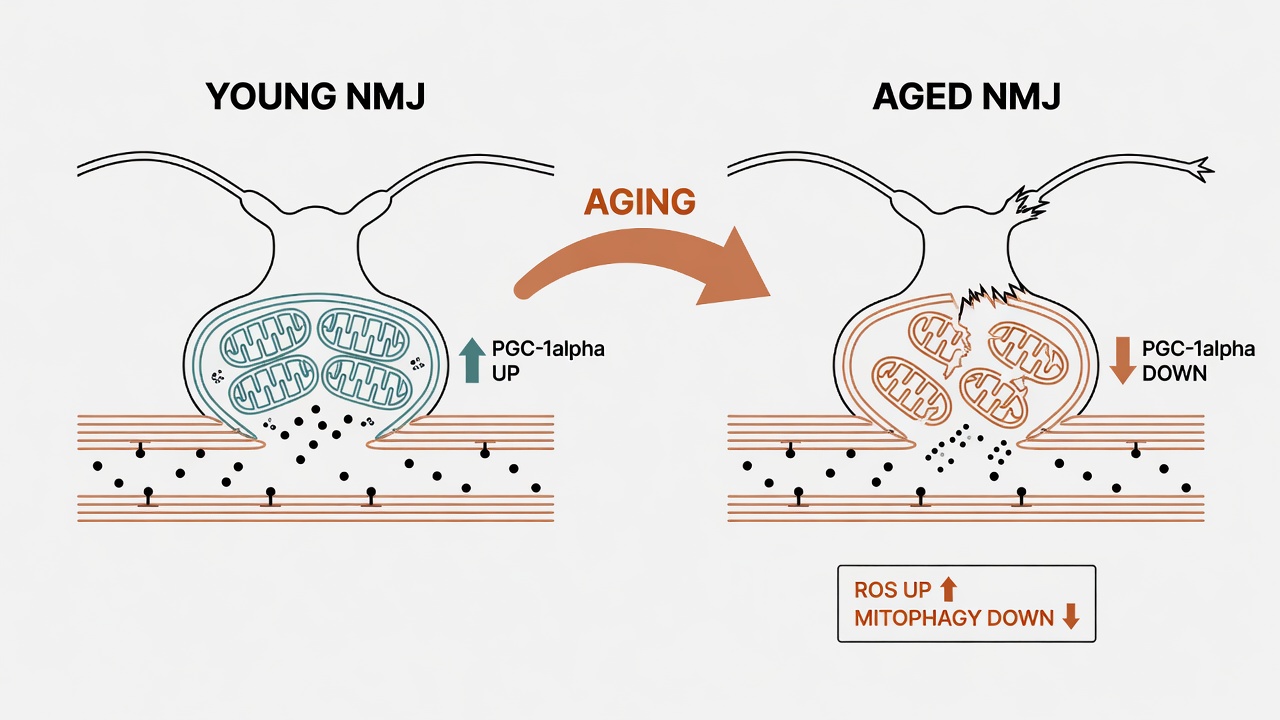
The NMJ's motor endplate is exquisitely sensitive to redox status. As mitochondrial function declines — ROS production rises, respiratory chain efficiency drops — the NMJ is among the first structures to absorb the impact. Declining expression of PGC-1alpha, the master regulator of mitochondrial biogenesis, disrupts fusion/fission balance and mitophagy, leading to NMJ fragmentation, denervation, and ultimately muscle weakness.
This is one of the molecular origins of sarcopenia. The "getting weaker with age" you feel is mitochondria collapsing at this most vulnerable outpost.
The most effective known intervention remains exercise. PGC-1alpha responds to mechanical loading more robustly than to any drug. Mitochondria-targeted peptides like SS-31 (elamipretide) are under investigation but lack sufficient dose-response data for clinical use.
Brain Aging Is Not a One-Way Street
While the NMJ raises the first red flag in the peripheral system, the hippocampus plays an analogous role in the central nervous system.
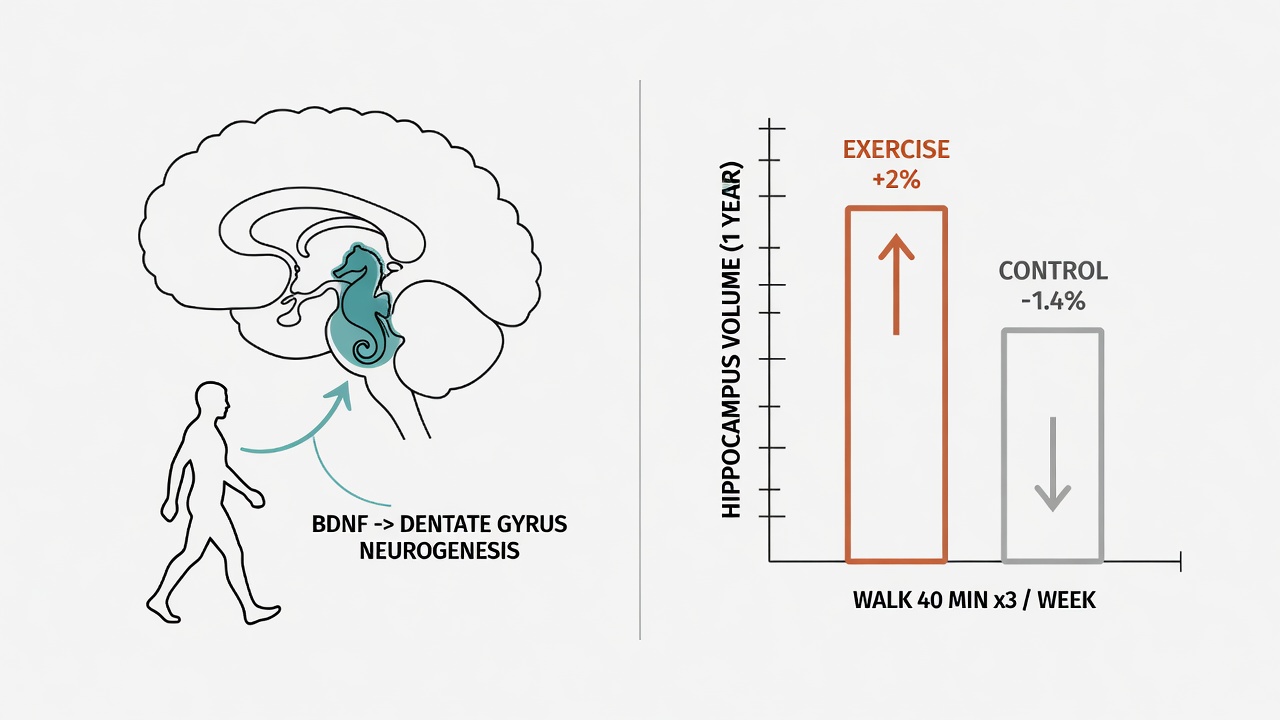
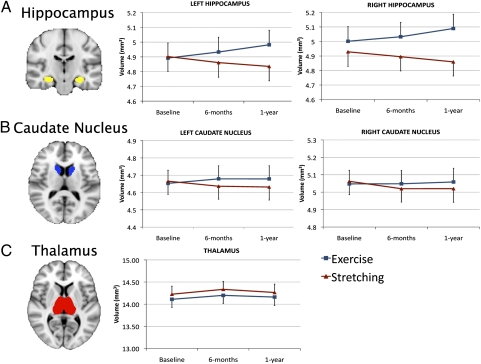
The hippocampus governs memory encoding and spatial navigation. It shrinks by roughly 1-2% per year with age, accompanied by cognitive decline, particularly in memory.
But a randomized controlled trial by Erickson et al. (2011, PNAS) delivered a striking signal: in adults aged 55-80, walking 40 minutes three times per week for one year increased hippocampal volume by approximately 2%, equivalent to reversing one to two years of natural atrophy. The control group shrank by 1.4% over the same period.
The mechanism involves BDNF (brain-derived neurotrophic factor). Aerobic exercise stimulates BDNF secretion, which promotes neurogenesis in the dentate gyrus — precisely the region that atrophies fastest with age and is earliest affected in Alzheimer's disease.
In other words, brain aging has a leverage point. It is not an irreversible one-way street.
Aging Is Cross-Organ Systems Engineering
If you thought aging is just individual organs deteriorating independently, a 2026 Cell Metabolism study may change your perspective.
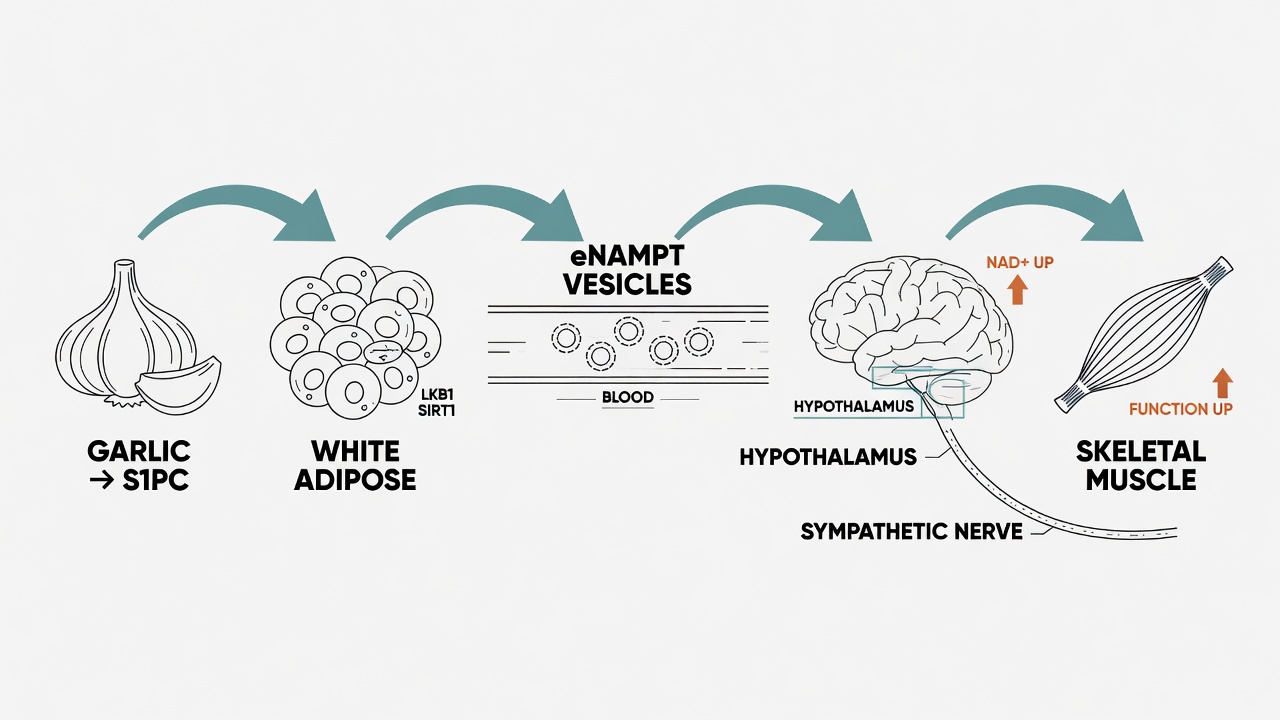
Researchers found that S1PC (S-1-propenyl-L-cysteine), a sulfur-containing metabolite from aged garlic extract, activates an unexpected cross-organ signaling axis: white adipose tissue, blood circulation, hypothalamus, skeletal muscle.
The pathway: S1PC promotes LKB1-STRAD-MO25 complex formation in adipocytes, activating LKB1. Activated LKB1 increases SIRT1 phosphorylation, driving adipose tissue to secrete extracellular vesicles containing eNAMPT. These eNAMPT vesicles enter circulation, preferentially target the hypothalamus, raise local NAD+ levels, and subsequently enhance skeletal muscle oxidative metabolism and contractile force via sympathetic nervous signaling (Suzuki et al., 2026).
In aged mice treated with S1PC from 15 to 23 months of age, frailty indices declined, body temperature recovered toward youthful levels, and grip strength improved — not through increased muscle mass, but through improved muscle functional quality.
The conceptual breakthrough: white adipose tissue is repositioned as an active endocrine/NAD+ vesicle relay station capable of remotely regulating brain-muscle aging physiology. Aging is not one organ breaking down alone — it is the inter-organ communication network gradually failing.
Human data remain preliminary: a randomized, double-blind, placebo-controlled acute study showed circulating eNAMPT elevation at 120 minutes in subjects over 40 with healthy body fat who consumed 25 mg S1PC. This is a translational biomarker signal, not a clinical endpoint.
What You Can Do: Survival Strategies Across Time
Understanding antagonistic pleiotropy yields one core insight: there are no eternally good strategies, only strategies executed at the right time.
Maintain your mitochondrial frontline. The NMJ is aging's earliest target, and PGC-1alpha is the core of the defense. Resistance training and aerobic exercise remain the most potent known stimuli for PGC-1alpha expression. This is not optional — it is necessary maintenance of basic physiological function.
Protect your hippocampus. Three sessions per week, 40 minutes of moderate-intensity walking, has RCT evidence for reversing hippocampal atrophy. The threshold is low, but consistency matters more than intensity.
Respect your biological clock. Circadian rhythm is not merely a sleep quality issue. It regulates hormone secretion, immune function, and metabolic efficiency. Stable routines, regular light exposure, and avoiding late-night eating — these seemingly mundane recommendations are backed by Nobel Prize-level molecular mechanisms.
Conclusion
"Longevity genes" gave us a false promise: as if somewhere in the genetic code sits a one-way ticket to a hundred. But genes are not static talismans. They are functions of time, playing different roles at different life stages — sometimes protecting you, sometimes working against you.
Aging is not uniform decline. It is a tug-of-war conducted simultaneously across multiple scales: molecular, cellular, organ, and inter-organ communication networks. The NMJ may start loosening in your forties, the hippocampus quietly shrinks year after year, and your adipose tissue is deciding whether to keep relaying messages to your brain and muscles.
The good news: this tug-of-war is not a spectator sport. You can hold your end of the rope.
References
- Williams GC (1957). Pleiotropy, natural selection, and the evolution of senescence. Evolution 11(4):398-411.
- Zehring WA, Wheeler DA, Reddy P, et al. (1984). P-element transformation with period locus DNA restores rhythmicity. Cell 39:369-376.
- Vosshall LB, Price JL, Sehgal A, et al. (1994). Block in nuclear localization of period protein by timeless. Science 263:1606-1609.
- Price JL, Blau J, Rothenfluh A, et al. (1998). double-time regulates PERIOD protein accumulation. Cell 94:83-95.
- Erickson KI, Voss MW, Prakash RS, et al. (2011). Exercise training increases size of hippocampus and improves memory. PNAS 108(7):3017-3022. doi: 10.1073/pnas.1015950108.
- Suzuki et al. (2026). S1PC activates an adipose-brain-muscle anti-aging axis via eNAMPT extracellular vesicles. Cell Metabolism. doi: 10.1016/j.cmet.2026.04.006.
- Chai S, Zhang N, Cui C, et al. (2025). Systematic review of mitochondrial dysfunction and oxidative stress in aging: A focus on neuromuscular junctions. Neural Regen Res 21(5):1947-1960. doi: 10.4103/NRR.NRR-D-24-01338.
Found this useful?
Follow for new AI × biomedical research notes:
Or buy me a coffee to keep new content coming.
☕ Buy Me a Coffee